Ants, Bees, Genomes & Evolution @ Queen Mary University London
Genome analyses for emerging model organisms
Using modern molecular tools on emerging (non-model) organism makes it possible to address exciting new questions. But the data aren’t as perfect they should be. In particular, genomes created from Roche 454, Illumina or ABI Solid sequence are fragmented: You wish you’d get a FASTA file with one long sequence per chromosome. Dream on! You get sequences for dozens to thousands of scaffolds. Each scaffold is a series of contigs, separated by stretches of unresolved NNNNNNNNN sequence (usually repetitive sequences). But the assembler knows these contigs are adjacent thanks to paired reads.
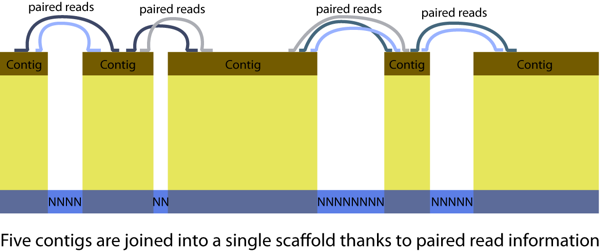
Genome fragmentation can make things challenging. Some tips from my experience with ant genomes:
How can you determine what is inside the unresolved poly-NNNNN sequence without genome walking or PCR and sequencing? Getting the whole thing will be difficult. But its easy to get a little:
-
Select resolved genomic sequence.
-
BLASTN against the raw unassembled reads (need help setting up a custom BLAST server?)
-
Among the reads that match your query, some will give you sequence that extends inside the unresolved NNNNNN region. If the sequence is known why wasn’t it shown? Because it’s repetitive nature made the sequence ambiguous for the assembly software.
**Are two scaffolds adjacent? **
-
Perhaps some paired reads do link them - but there were insufficient data for the assembler to be sure. With 454 assemblies, check newbler’s output in the 454Scaffolds.txt and _454PairStatus.txt _files. These report where all paired reads map.
-
Or map independently obtained transcriptome or proteome data onto your genome. Oksana Riba-Grognuz developed an easy way of visualizing RNA mapped to genomes.
-
Or check a closely related species - perhaps the region is better assembled there.
How good is good enough? Some sequence/data/scaffolds/models are missing or mediocre! But no biological dataset is ever perfect. If you’re trying to make your emerging model organism’s data perfect… you’ll get nowhere fast. The 20% effort that bring you 80% of the way will probably be good enough to answer your exciting biological question.
September 21, 2011